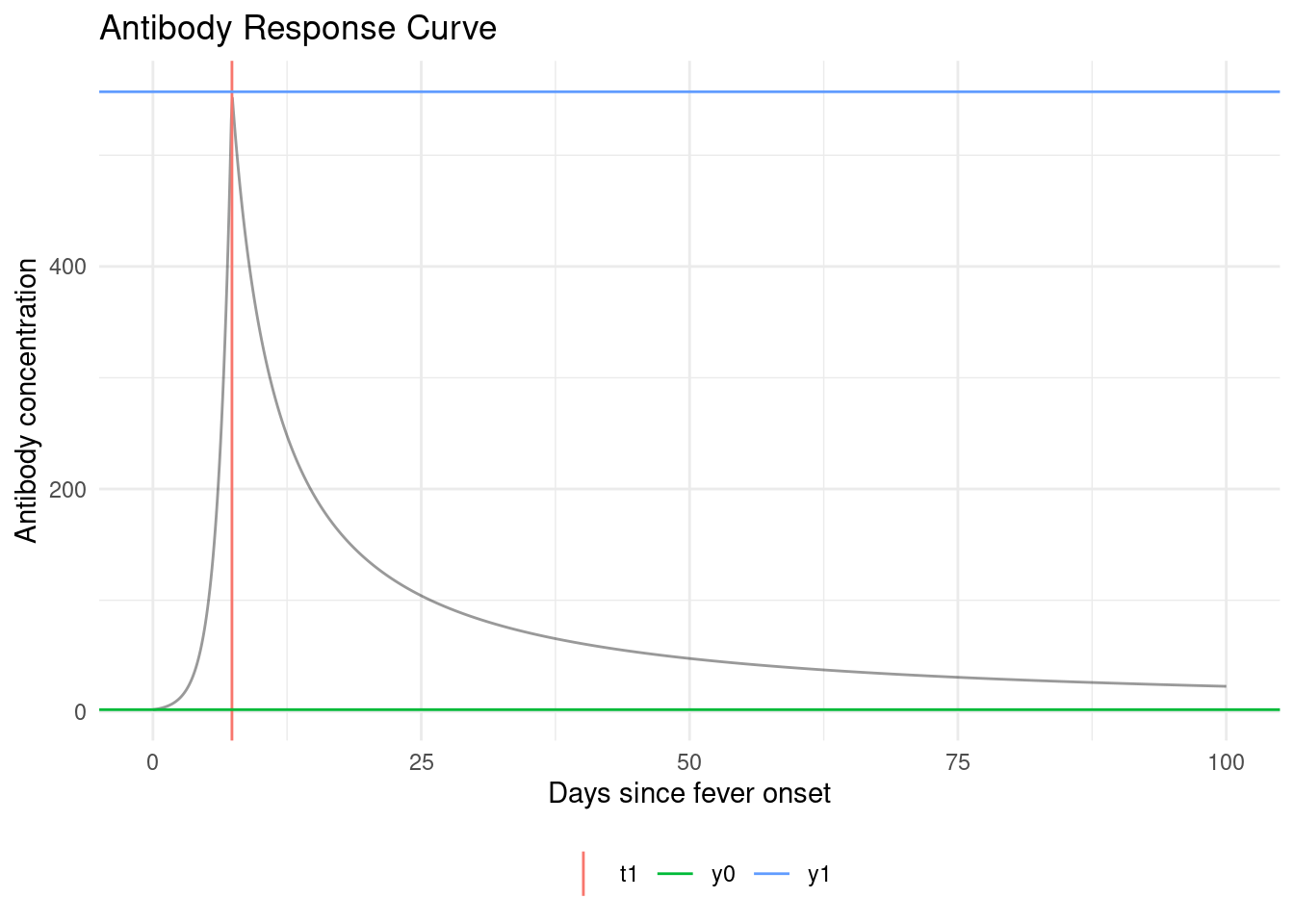
Unfortunately, we don’t observe infection times ; we only observe antibody levels . So things get a little more complicated.
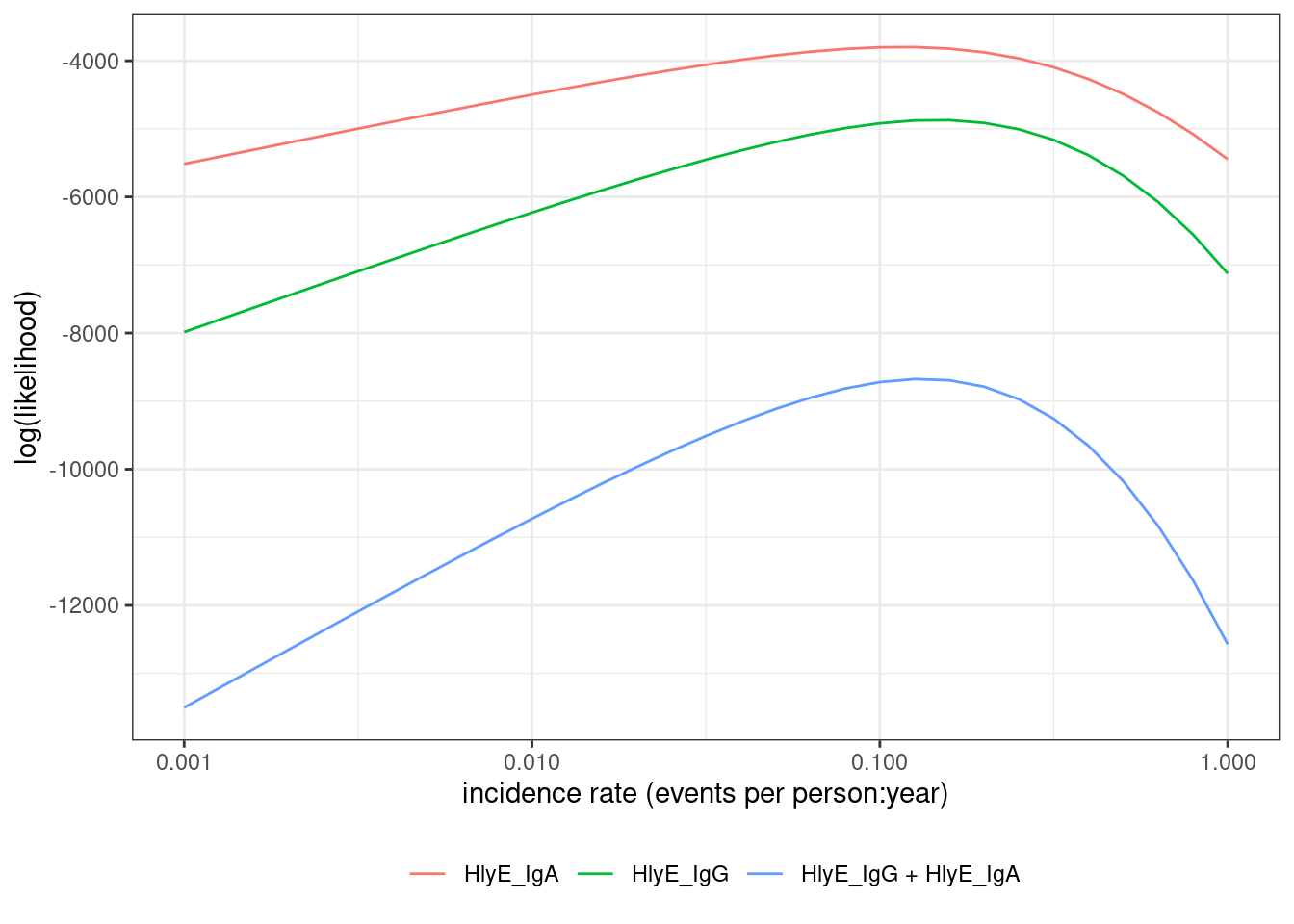
In short, we are hoping that we can estimate (time since last infection) from (current antibody levels). If we could do that, then we could plug in our estimates into that likelihood above, and estimate as previously.
We’re actually going to do something a little more nuanced; instead of just using one value for , we are going to consider all possible values of for each individual.
We need to link the data we actually observed to the incidence rate.
The likelihood of an individual’s observed data, , can be expressed as an integral over the joint likelihood of and (using the Law of Total Probability):